read.table() uses quote="'\""
(that is, looking for either single- or double-quotes)
read.delim() uses quote="\"" (just looking for double-quotes).To solve quote problem,
In
read.table(), use quote="", and for that matter also fill=FALSE
read.table() uses quote="'\""
(that is, looking for either single- or double-quotes)
read.delim() uses quote="\"" (just looking for double-quotes).read.table(), use quote="", and for that matter also fill=FALSE
X column1 column2 column3
row1 0 1 2
row2 3 4 5
row3 6 7 8
row4 9 10 11
to
X row1 row2 row3 row4
column1 0 3 6 9
column2 1 4 7 10
column3 2 5 8 11 A. awk '
{
for (i=1; i<=NF; i++) {
a[NR,i] = $i
}
}
NF>p { p = NF }
END {
for(j=1; j<=p; j++) {
str=a[1,j]
for(i=2; i<=NR; i++){
str=str" "a[i,j]; # check FS (ex: space, tab, etc)
}
print str
}
}' filep = rand(10,2);
scatter(p(:,1), p(:,2), 'filled')
axis([0 1 0 1])
labels = num2str((1:size(p,1))','%d'); %'
text(p(:,1), p(:,2), labels, 'horizontal','left', 'vertical','bottom','Fontsize', 7)scp <source> <destination>
B to A while logged into B:scp /path/to/file username@a:/path/to/destination
B to A while logged into A:scp username@b:/path/to/file /path/to/destination


tar zxvf cmake-3.*
cd cmake-3.*
./bootstrap --prefix=/usr/local
make
make install cmake --version
cmake version *.*.*
CMake suite maintained and supported by Kitware (kitware.com/cmake). sudo rpm –i sample_file.rpmsudo yum localinstall sample_file.rpm sudo rpm –e sample_file.rpm grep -v -e foo -e bar filegrep -Ev 'word1|word2'
Substring Extraction${string:position}Extracts substring from$stringat$position.If the$stringparameter is "*" or "@", then this extracts the positional parameters, starting at$position.${string:position:length}Extracts$lengthcharacters of substring from$stringat$position.
stringZ=abcABC123ABCabc
# 0123456789.....
# 0-based indexing.
echo ${stringZ:0} # abcABC123ABCabc
echo ${stringZ:1} # bcABC123ABCabc
echo ${stringZ:7} # 23ABCabc
echo ${stringZ:7:3} # 23A
# Three characters of substring.
# Is it possible to index from the right end of the string?
echo ${stringZ:-4} # abcABC123ABCabc
# Defaults to full string, as in ${parameter:-default}.
# However . . .
echo ${stringZ:(-4)} # Cabc
echo ${stringZ: -4} # Cabc
# Now, it works.
# Parentheses or added space "escape" the position parameter.samtools view -h input.indexed.bam "Chr1:10000-20000" > output.sam
numpy.isin(element, test_elements, assume_unique=False, invert=False)[source]| Parameters: |
|
|---|---|
| Returns: |
|
comm <(sort a.txt) <(sort b.txt)
$sed 's/apple/lemon/g' test.txt
some_command >file.log 2>&1 fit <- lmFit(myVoomData,design)
fit <- eBayes(fit)
options(digits=3)
writefile = topTable(fit,n=Inf,sort="none", p.value=0.01)
write.csv(writefile, file="file.csv")
My problem is that all of the 6156 genes are differentially expressed (p-value 0.01). Only a few hundred were differentially expressed using DESe2, but I guess that can't be trusted.eBayes with robust=TRUE as well as trend=TRUE. That will reduce the sensitivity to zero variances.
-v option: (P.S. use a space after -v or it will be less portable. E.g., awk -v var= not awk -vvar=)variable="line one\nline two"
awk -v var="${variable}" 'BEGIN {print var}'
line one
line two awk and variable is available in the BEGIN block as well:awk -v a="${var1}" -v b="${var2}" 'BEGIN {print a,b}'os.system("some_command with args") passes the command and arguments to your system's shell. This is nice because you can actually run multiple commands at once in this manner and set up pipes and input/output redirection. For example: os.system("some_command < input_file | another_command > output_file") stream = os.popen("some_command with args") will do the same thing as os.system except that it gives you a file-like object that you can use to access standard input/output for that process. There are 3 other variants of popen that all handle the i/o slightly differently. If you pass everything as a string, then your command is passed to the shell; if you pass them as a list then you don't need to worry about escaping anything. See the documentation.Popen class of the subprocess module. This is intended as a replacement for os.popen but has the downside of being slightly more complicated by virtue of being so comprehensive. For example, you'd say:print subprocess.Popen("echo Hello World", shell=True, stdout=subprocess.PIPE).stdout.read()print os.popen("echo Hello World").read()call function from the subprocess module. This is basically just like the Popen class and takes all of the same arguments, but it simply waits until the command completes and gives you the return code. For example:return_code = subprocess.call("echo Hello World", shell=True) subprocess.run function, which is a lot like the above but even more flexible and returns a CompletedProcess object when the command finishes executing.subprocess module should probably be what you use.print subprocess.Popen("echo %s " % user_input, stdout=PIPE).stdout.read()
| ||||||||||||||||||||||||||||||||||
ChIP-seq guidelines and practices of the ENCODE and modENCODE consortiahttps://genome.cshlp.org/content/22/9/1813.longRecommended Coverage and Read Depth for NGS Applicationshttps://genohub.com/recommended-sequencing-coverage-by-application/Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data | ||||||||||||||||||||||||||||||||||
| https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003326 | ||||||||||||||||||||||||||||||||||
Clear description for normalization method and DESeq2 normalization process (recommend)
https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html
Gene expression units explained: RPM, RPKM, FPKM, TPM, DESeq, TMM, SCnorm, GeTMM, and ComBat-Seq : include python, R code (recommend)
https://www.reneshbedre.com/blog/expression_units.html
calculation of CPM, TPM, FPKM ; R code (recommend)
https://haroldpimentel.wordpress.com/2014/05/08/what-the-fpkm-a-review-rna-seq-expression-units/
How to normalized TPM with TMM method?
https://www.biostars.org/p/388584/
->you have TMM factors and want to compute TPMs.
TMM factors can in principle be used to compute TPMs. In edgeR, any downstream quantity that is computed from the library sizes will incorporate the TMM factors automatically, because the factors are considered part of the effective library sizes. TMM normalization factors will be applied automatically when you use
CPM <- cpm(dge)
or
RPKM <- rpkm(dge)
in edgeR to compute CPMs or RPKMs from a DGEList object. I don't necessarily recommend TPM values myself, but if you go on to compute TPMs by
TPM <- t( t(RPKM) / colSums(RPKM) ) * 1e6
then the TMM factors will naturally have been incorporated into the computation.
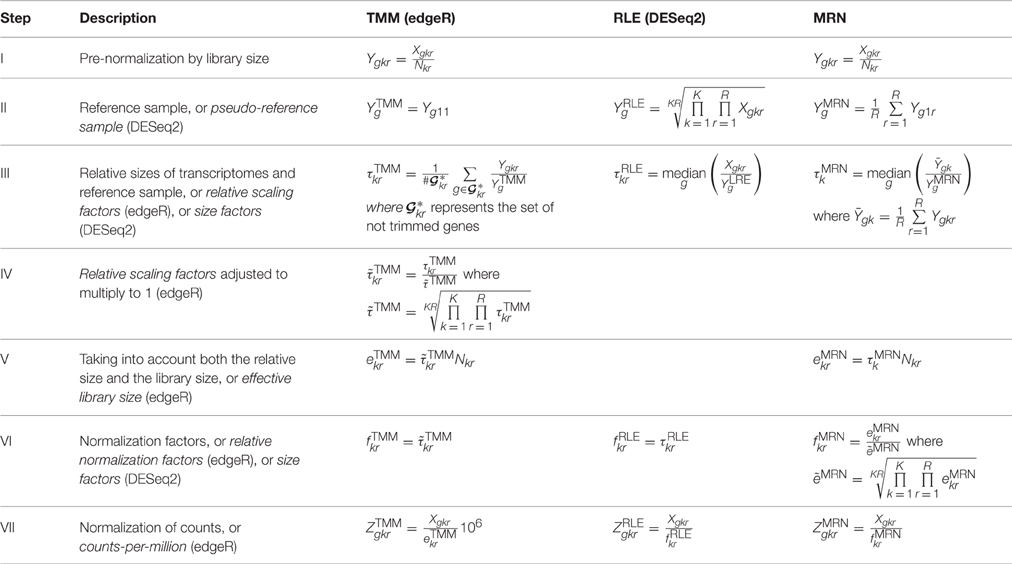
TMM (edgeR), RLE (DESeq2), and MRN Normalization Methods comparison
https://www.frontiersin.org/articles/10.3389/fgene.2016.00164/full