Tophat2 is not bad
http://cdwscience.blogspot.com/2019/01/tophat-really-isnt-that-bad.html
https://genomespot.blogspot.com/2015/03/hisat-vs-star-vs-tophat2-vs-olego-vs.html
Tophat2 is bad
Question: HISAT2 or Tophat2
https://www.biostars.org/p/230724/
May 22, 2020
Apr 8, 2020
bedops --chop : chop bed into identical intervals
https://bedops.readthedocs.io/en/latest/content/reference/set-operations/bedops.html
The
--chop
operator merges all overlapping input regions and “chops” them up into a
set of disjoint segments of identical length (with a default of one
base). One or more input files may be provided; this option will segment
regions from all inputs:
Note
Overlapping and nested regions are merged into
contiguous ranges before chopping. The end result contains unique,
non-overlapping elements.
chr1 0 30427671
chr2 0 19698289
chr3 0 23459830
chr4 0 18585056
chr5 0 26975502
(base) [lee@ko44 annotation]$ bedops --chop 1000 chrom.sizes.chr.TAIR10.bed > arabidopsis_1kb_chr1_5.bed
(base) [lee@ko44 annotation]$ more arabidopsis_1kb_chr1_5.bed
chr1 0 1000
chr1 1000 2000
chr1 2000 3000
chr1 3000 4000
chr1 4000 5000
chr1 5000 6000
chr1 6000 7000
chr1 7000 8000
chr1 0 1000
chr1 1000 2000
chr1 2000 3000
chr1 3000 4000
chr1 4000 5000
chr1 5000 6000
chr1 6000 7000
chr1 7000 8000
Mar 31, 2020
R for Data Science
https://r4ds.had.co.nz/

how to get your data into R, get it into the most useful structure, transform it, visualise it and model it.
how to get your data into R, get it into the most useful structure, transform it, visualise it and model it.
Mar 25, 2020
changing chromosome notation in .BAM file
http://seqanswers.com/forums/showthread.php?t=22504
https://josephcckuo.wordpress.com/2016/11/17/modify-chromosome-notation-in-bam-file/
change chromosome notation "1" to "chr1"
for file in *.bam; do filename=`echo $file | cut -d "." -f 1`; samtools view -H $file | sed -e 's/SN:\([0-9XY]\)/SN:chr\1/' -e 's/SN:MT/SN:chrM/' | samtools reheader - $file > ${filename}_chr.bam; done
for loop
for file in *.bam; do filename=`echo $file | cut -d "." -f 1`; samtools view -H $file | sed -e 's/SN:\([0-9XY]\)/SN:chr\1/' -e 's/SN:MT/SN:chrM/' | samtools reheader - $file > ${filename}_chr.bam; done
https://josephcckuo.wordpress.com/2016/11/17/modify-chromosome-notation-in-bam-file/
change chromosome notation "1" to "chr1"
for file in *.bam; do filename=`echo $file | cut -d "." -f 1`; samtools view -H $file | sed -e 's/SN:\([0-9XY]\)/SN:chr\1/' -e 's/SN:MT/SN:chrM/' | samtools reheader - $file > ${filename}_chr.bam; done
for loop
for file in *.bam; do filename=`echo $file | cut -d "." -f 1`; samtools view -H $file | sed -e 's/SN:\([0-9XY]\)/SN:chr\1/' -e 's/SN:MT/SN:chrM/' | samtools reheader - $file > ${filename}_chr.bam; done
Mar 24, 2020
deepTools: tools for exploring deep sequencing data
https://deeptools.readthedocs.io/en/latest/index.html
deepTools is a suite of python tools particularly developed for the efficient analysis of high-throughput sequencing data, such as ChIP-seq, RNA-seq or MNase-seq.
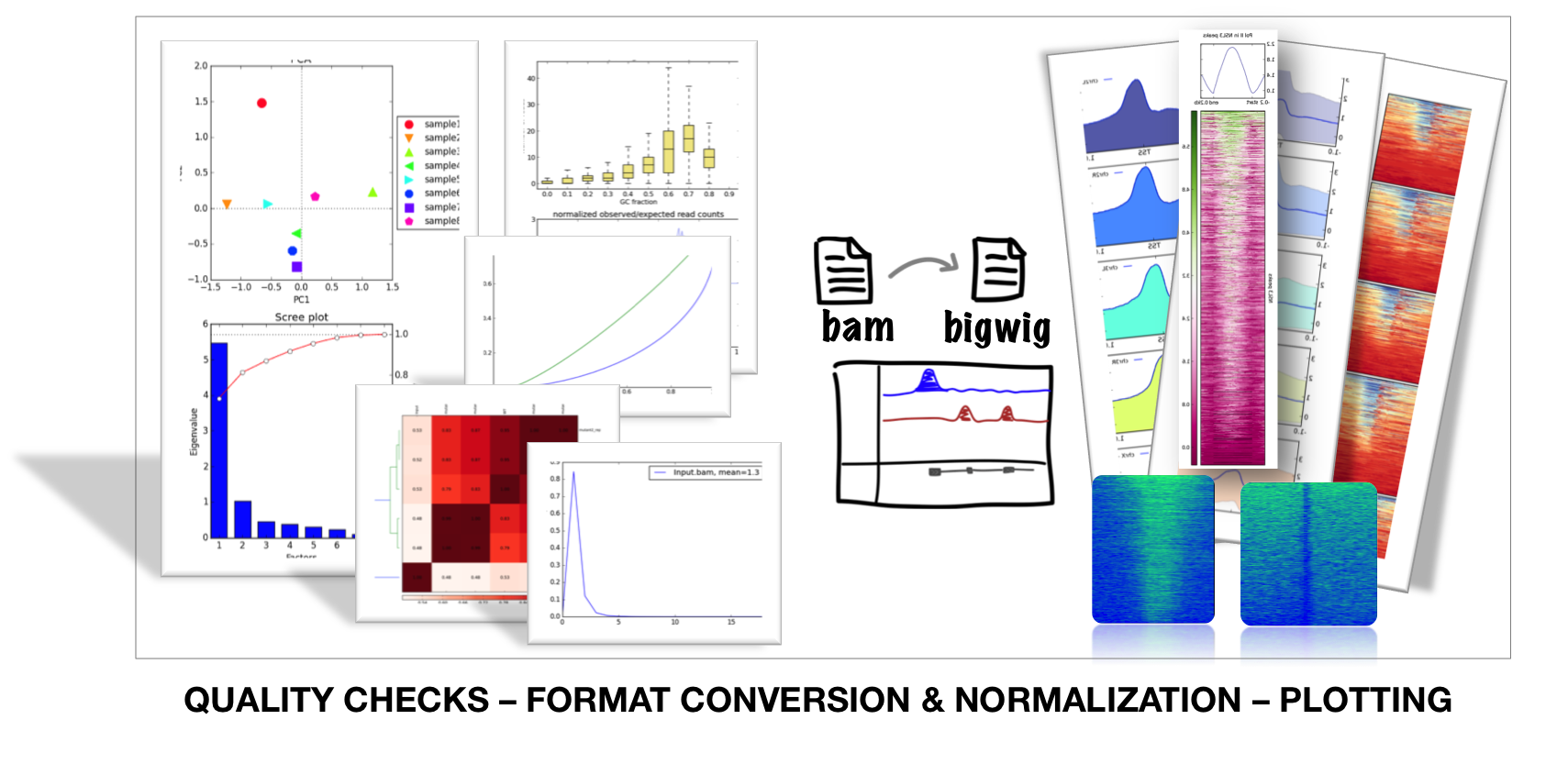

deepTools is a suite of python tools particularly developed for the efficient analysis of high-throughput sequencing data, such as ChIP-seq, RNA-seq or MNase-seq.
Mar 4, 2020

SeqPlots : interactive tool for visualizing track signals and sequence motif densities along genomic features using average plots and heatmaps
https://github.com/Przemol/seqplots
SeqPlots is a web browser tool for plotting average track signals (e.g. read coverage) and sequence motif densities over user-specified genomic features.
Standalone versions of SeqPlots are available as a Mac OS X (10.6 or higher) app bundle combing R, all required packages and scripts([subproject home]) or as an R package ([subproject home]).
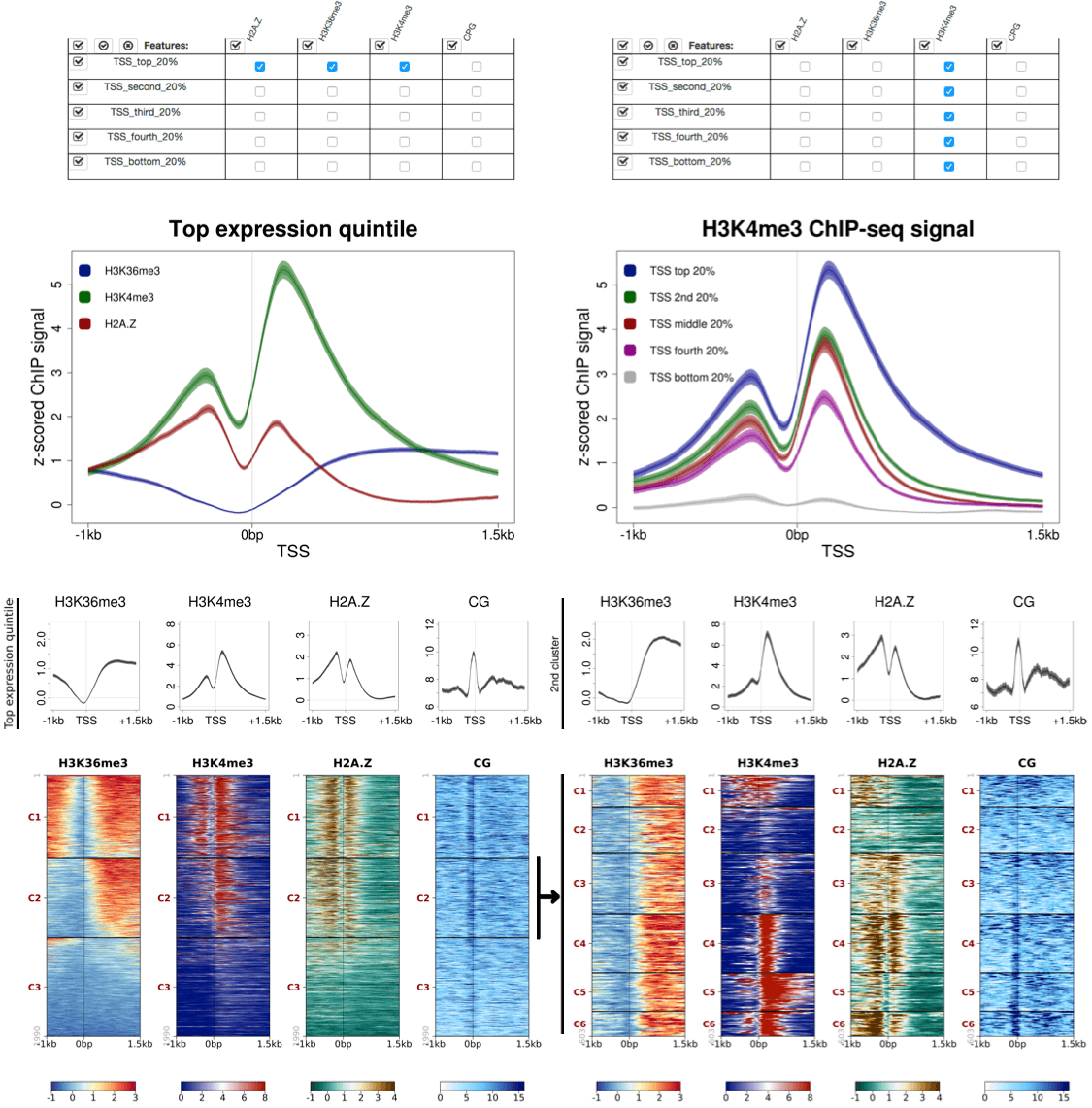
SeqPlots is a web browser tool for plotting average track signals (e.g. read coverage) and sequence motif densities over user-specified genomic features.
Standalone versions of SeqPlots are available as a Mac OS X (10.6 or higher) app bundle combing R, all required packages and scripts([subproject home]) or as an R package ([subproject home]).
Subscribe to:
Posts (Atom)