clear description of fastQC read duplication report and example
https://www.biostars.org/p/107402/
Jul 5, 2019
May 31, 2019
Differential expression of FPKM from RNA-seq data using limma and voom()
Differential expression of FPKM from RNA-seq data using limma and voom()
https://support.bioconductor.org/p/56275/
Q.
A.
No, it is absolutely not ok to input FPKM values to voom. The results will be nonsense. Same goes for edgeR and DESeq, for the reasons explained in the documentation for those packages.
Note that a matrix of FPKM is not a matrix of counts.
It seems to me that you have three options in decreasing order of desirability:
What you're doing wrong is trying to do a DE analysis using FPKMs in the first place. There is no good answer to what the FPKM offset should be to avoid taking the log of zero, which is one of the problems with using FPKMs. I would personally use a larger offset than any you have used, certainly not less than 0.1, but then I wouldn't do it at all.
The way to choose a tuning parameter like this is to plot the data to see that it looks vaguely reasonable rather than to count the number of DE genes.
I assume you have filtered the data so that you are not trying to analyse genes for which all, or almost all, the FPKM are zero. Having lots of FPKM=0 would lead to the warnings about zero variances.
I also suggest you used
https://support.bioconductor.org/p/56275/
Q.
I have a count matrix of FPKM values and I want to estimate differentially expressed genes between two conditions. First I used DESeq2, but I realized that this is not good for FPKM values. I then transformed the counts using voom() in the limma package and then used:
I am new to this type of analysis, and to R, but is it ok to simply transform the data by voom()? Can I use the transformed data in DESeq2? Any other ways I can use FPKM counts to estimate differentially expressed genes?
fit <- lmFit(myVoomData,design)
fit <- eBayes(fit)
options(digits=3)
writefile = topTable(fit,n=Inf,sort="none", p.value=0.01)
write.csv(writefile, file="file.csv")
My problem is that all of the 6156 genes are differentially expressed (p-value 0.01). Only a few hundred were differentially expressed using DESe2, but I guess that can't be trusted.I am new to this type of analysis, and to R, but is it ok to simply transform the data by voom()? Can I use the transformed data in DESeq2? Any other ways I can use FPKM counts to estimate differentially expressed genes?
A.
No, it is absolutely not ok to input FPKM values to voom. The results will be nonsense. Same goes for edgeR and DESeq, for the reasons explained in the documentation for those packages.
Note that a matrix of FPKM is not a matrix of counts.
It seems to me that you have three options in decreasing order of desirability:
- Get the actual integer counts from which the FPKM were computed and do a proper analysis, for example using voom or edgeR.
- Get the gene lengths and library sizes used to compute the FPKM and convert the FPKM back to counts.
- If FPKM is really all you have, then convert the values to a log2 scale (y = log2(FPKM+0.1) say) and do an ordinary limma analysis as you would for microarray data, using eBayes() with trend=TRUE. Do not use voom, do not use edgeR, do not use DESeq. (Do not pass go and do not collect $200.) This isn't 100% ideal, but is probably the best analysis available.
What you're doing wrong is trying to do a DE analysis using FPKMs in the first place. There is no good answer to what the FPKM offset should be to avoid taking the log of zero, which is one of the problems with using FPKMs. I would personally use a larger offset than any you have used, certainly not less than 0.1, but then I wouldn't do it at all.
The way to choose a tuning parameter like this is to plot the data to see that it looks vaguely reasonable rather than to count the number of DE genes.
I assume you have filtered the data so that you are not trying to analyse genes for which all, or almost all, the FPKM are zero. Having lots of FPKM=0 would lead to the warnings about zero variances.
I also suggest you used
eBayes with robust=TRUE as well as trend=TRUE. That will reduce the sensitivity to zero variances.
How do I use shell variables in an awk script?
How do I use shell variables in an awk script?
https://stackoverflow.com/questions/19075671/how-do-i-use-shell-variables-in-an-awk-script
If you have multiple variables:
https://stackoverflow.com/questions/19075671/how-do-i-use-shell-variables-in-an-awk-script
- Using -v (The best way, most portable)
It uses the-v option: (P.S. use a space after -v or it will be less portable. E.g., awk -v var= not awk -vvar=)variable="line one\nline two"
awk -v var="${variable}" 'BEGIN {print var}'
line one
line two awk and variable is available in the BEGIN block as well:If you have multiple variables:
awk -v a="${var1}" -v b="${var2}" 'BEGIN {print a,b}'Calling an external command in Python - subprocess, os.system
How can I call an external command (as if I'd typed it at the Unix shell or Windows command prompt) from within a Python script?
https://stackoverflow.com/questions/89228/calling-an-external-command-in-python/89243#
Here's a summary of the ways to call external programs and the advantages and disadvantages of each:
Finally please be aware that for all methods where you pass the final command to be executed by the shell as a string and you are responsible for escaping it. There are serious security implications if any part of the string that you pass can not be fully trusted. For example, if a user is entering some/any part of the string. If you are unsure, only use these methods with constants.
To give you a hint of the implications consider this code:
os.system("some_command with args")passes the command and arguments to your system's shell. This is nice because you can actually run multiple commands at once in this manner and set up pipes and input/output redirection. For example:
However, while this is convenient, you have to manually handle the escaping of shell characters such as spaces, etc. On the other hand, this also lets you run commands which are simply shell commands and not actually external programs. See the documentation.os.system("some_command < input_file | another_command > output_file")
stream = os.popen("some_command with args")will do the same thing asos.systemexcept that it gives you a file-like object that you can use to access standard input/output for that process. There are 3 other variants of popen that all handle the i/o slightly differently. If you pass everything as a string, then your command is passed to the shell; if you pass them as a list then you don't need to worry about escaping anything. See the documentation.
- The
Popenclass of thesubprocessmodule. This is intended as a replacement foros.popenbut has the downside of being slightly more complicated by virtue of being so comprehensive. For example, you'd say:
instead of:print subprocess.Popen("echo Hello World", shell=True, stdout=subprocess.PIPE).stdout.read()
but it is nice to have all of the options there in one unified class instead of 4 different popen functions. See the documentation.print os.popen("echo Hello World").read() - The
callfunction from thesubprocessmodule. This is basically just like thePopenclass and takes all of the same arguments, but it simply waits until the command completes and gives you the return code. For example:
See the documentation.return_code = subprocess.call("echo Hello World", shell=True)
- If you're on Python 3.5 or later, you can use the new
subprocess.runfunction, which is a lot like the above but even more flexible and returns aCompletedProcessobject when the command finishes executing.
- The os module also has all of the fork/exec/spawn functions that you'd have in a C program, but I don't recommend using them directly.
subprocess module should probably be what you use.Finally please be aware that for all methods where you pass the final command to be executed by the shell as a string and you are responsible for escaping it. There are serious security implications if any part of the string that you pass can not be fully trusted. For example, if a user is entering some/any part of the string. If you are unsure, only use these methods with constants.
To give you a hint of the implications consider this code:
print subprocess.Popen("echo %s " % user_input, stdout=PIPE).stdout.read()ChIP-seq : read coverage and input
What is the minimum million reads that I need to have a good result of chip-seq in human?
Coverage depth recommend
https://www.researchgate.net/post/What_is_the_minimum_million_reads_that_I_need_to_have_a_good_result_of_chip-seq_in_human
What matters is unique reads rather than total reads. ChIP-Seq runs that give 20-30 million unique reads will work quite well for most factors.
How many input samples do you need to add to ChIP-Seq library?
https://www.researchgate.net/post/How_many_input_samples_do_you_need_to_add_to_ChIP-Seq_library
best control is the input split from the IP reaction, but given economic constraints I would argue a pooled input will (on the whole) give similar results.
Question about inputs for ChIP-seq
http://seqanswers.com/forums/showthread.php?t=35377
Coverage depth recommend
https://www.illumina.com/science/education/sequencing-coverage.htm
Sequencing Coverage Calculator
https://support.illumina.com/downloads/sequencing_coverage_calculator.html
Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP)
Nature Protocols volume 5, pages 457–472 (2010)
https://www.nature.com/articles/nprot.2009.244
| ||||||||||||||||||||||||||||||||||
ChIP-seq guidelines and practices of the ENCODE and modENCODE consortiahttps://genome.cshlp.org/content/22/9/1813.longRecommended Coverage and Read Depth for NGS Applicationshttps://genohub.com/recommended-sequencing-coverage-by-application/Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data | ||||||||||||||||||||||||||||||||||
| https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003326 | ||||||||||||||||||||||||||||||||||
How to tell which library type to use in bowtie, tophat (fr-unstrand, -firststrand -secondstrand)?
How to tell which library type to use (fr-unstrand, -firststrand or secondstrand)?
Normalization method of DESeq2, CPM, TPM, FPKM, TMM
Clear description for normalization method and DESeq2 normalization process (recommend)
https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html
Gene expression units explained: RPM, RPKM, FPKM, TPM, DESeq, TMM, SCnorm, GeTMM, and ComBat-Seq : include python, R code (recommend)
https://www.reneshbedre.com/blog/expression_units.html
calculation of CPM, TPM, FPKM ; R code (recommend)
https://haroldpimentel.wordpress.com/2014/05/08/what-the-fpkm-a-review-rna-seq-expression-units/
How to normalized TPM with TMM method?
https://www.biostars.org/p/388584/
->you have TMM factors and want to compute TPMs.
TMM factors can in principle be used to compute TPMs. In edgeR, any downstream quantity that is computed from the library sizes will incorporate the TMM factors automatically, because the factors are considered part of the effective library sizes. TMM normalization factors will be applied automatically when you use
CPM <- cpm(dge)
or
RPKM <- rpkm(dge)
in edgeR to compute CPMs or RPKMs from a DGEList object. I don't necessarily recommend TPM values myself, but if you go on to compute TPMs by
TPM <- t( t(RPKM) / colSums(RPKM) ) * 1e6
then the TMM factors will naturally have been incorporated into the computation.
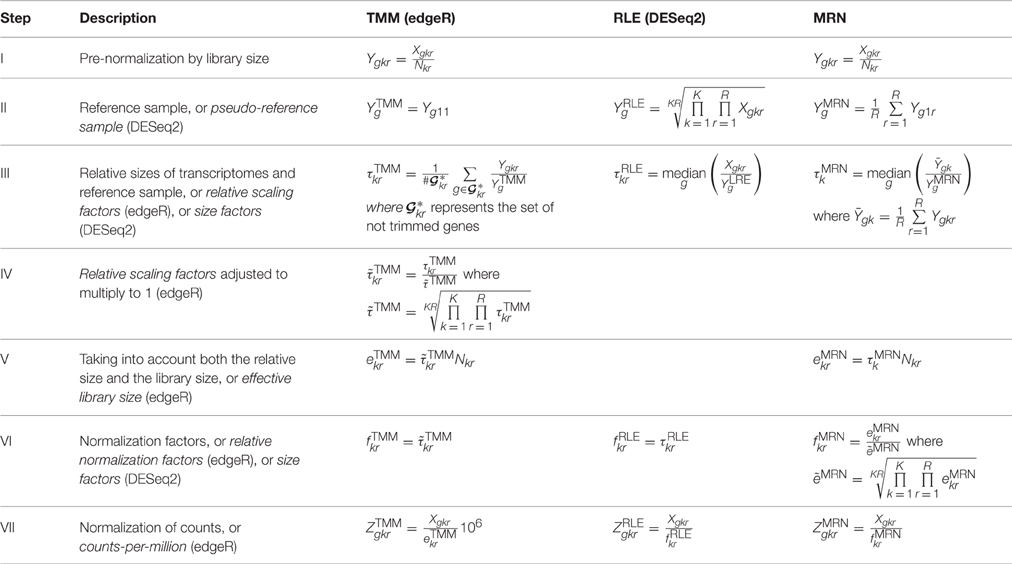
TMM (edgeR), RLE (DESeq2), and MRN Normalization Methods comparison
https://www.frontiersin.org/articles/10.3389/fgene.2016.00164/full
Subscribe to:
Posts (Atom)